Key Findings
A seminal research paper has been published on a formal theory for crystal structure prediction, referencing the inverse design of crystal structures using Constrained Crystal Deep Convolutional Generative Adversarial Networks (CG-DCGANs) and applications to material property prediction using Graph Neural Networks (GNNs). This theory promises to bring unprecedented efficiency and accuracy to the design of new materials with specific functionalities, further enhancing the role of AI in materials science, and is poised to accelerate discovery.
Technical Details and Mechanisms
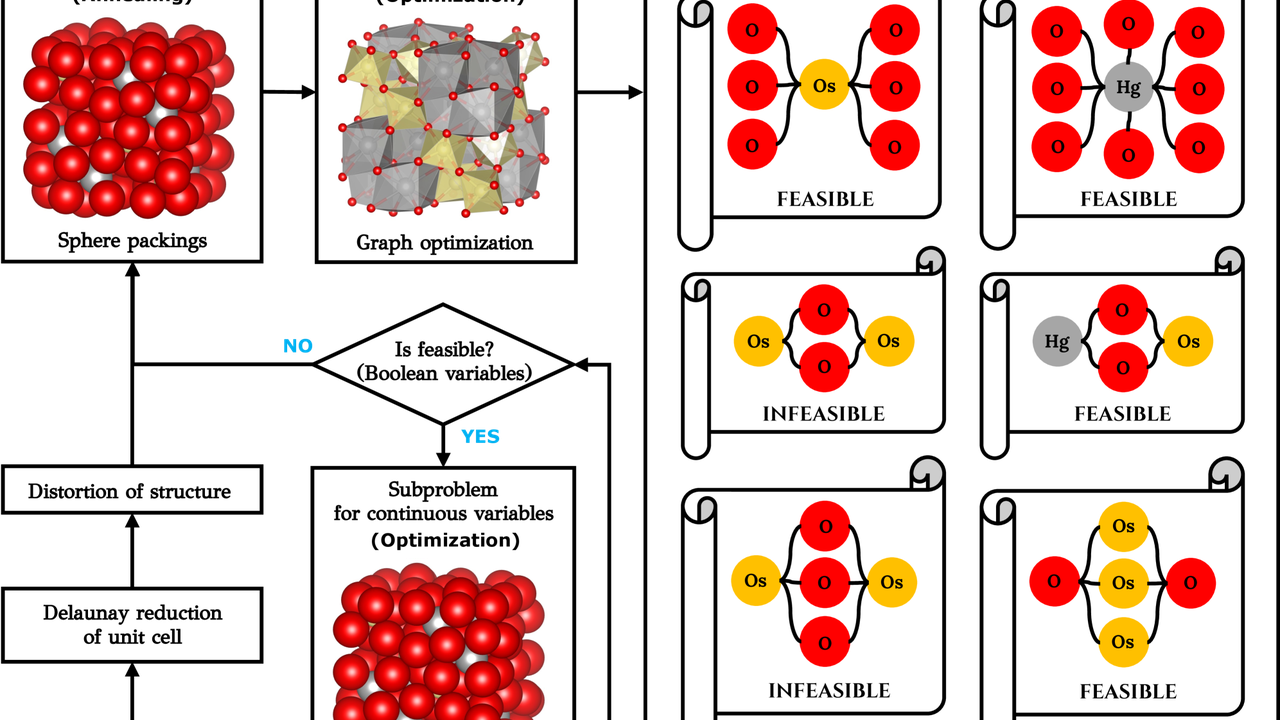
Crystal structure is a fundamental determinant of a material’s physical and chemical properties, making its prediction a central challenge in new material development. However, the search space for possible crystal structures is vast, and high-fidelity methods like first-principles calculations are computationally expensive. The formal theory proposed in this paper is innovative in several aspects:
- Inverse Design via Generative Models: CG-DCGANs generate new crystal structures (inverse design) with desired properties from learned data distributions of existing crystal structures. This opens the path from ‘properties to structure,’ contrasting with the traditional ‘composition to structure’ approach. This enables direct design of materials to meet specific functional requirements such as electrical conductivity, thermal conductivity, or mechanical strength.
- Property Prediction via Graph Neural Networks (GNNs): GNNs represent crystal structures as graphs and learn/predict material properties from interatomic bonds and geometric arrangements. This effectively captures complex material interactions, allowing for rapid and high-accuracy property evaluation.
- Integration with Mathematical Optimization: The proposed theory incorporates Generalized Disjunctive Programming, allowing physical constraints such as crystal space group symmetry to be directly embedded into the design process. This ensures that the generated structures are physically stable and experimentally feasible.
These technologies bridge the gap between ‘experiment’ and ‘theory’ in computational materials science, significantly shortening the design-to-realization cycle.
Industry Context and Future Outlook
The advancements in this formal theory and associated AI technologies hold the potential to accelerate the discovery and development of groundbreaking new materials in fields such as semiconductors, batteries, superconductors, and catalysts. For instance, highly efficient solar cell materials, ultra-low-loss data transmission materials, and innovative pharmaceutical intermediates could be designed in much shorter timeframes than ever before. The ability to inversely design crystal structures is expected to shift the R&D strategy from ‘exploration’ to ‘design,’ fundamentally transforming the competitive landscape in materials innovation. This research marks a significant milestone in demonstrating how computational materials science will advance to its next frontier driven by AI.








Comments